

????????????????Process validation for start-up medical device companies is essential to ensure with confidence, and a high degree of certainty, that their manufacturing processes have the proper application and controls in place to produce products that are safe and meet the specifications, quality and user requirements.
If you?re on a fast-track plan to implement your ISO 13485:2016 quality system, you should not take any shortcuts with your process validations. As a start-up company you will likely not have a long history with your product design or required manufacturing processes. If this is the case in my experience, it is far better to overdo the degree of validation rather that under perform.
The US FDA Quality System Regulation defines process validation as:
Process validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications?
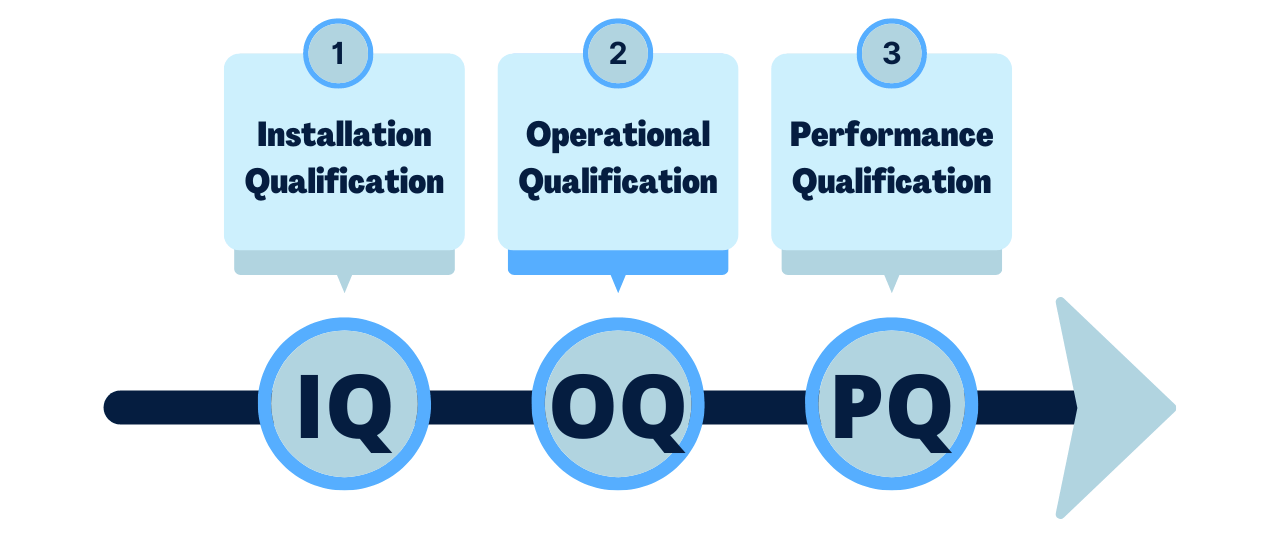
In this article we will focus on the 3 basic steps in the validation process, IQ, OQ, and PQ.
Will also cover briefly, Master Validation Plans, Factory Acceptance Tests, and Software Validation and will cover those in more detail in future posts.
Before we go into more detail on the validation process let?s take a look at the regulatory compliance requirements in ISO 13485:2016 and the FDA regulations.
Validation requirements to meet ISO 13485:2016 & FDA
ISO 13485:2016 7.5.6 Validation of processes for production and service provision
The organization shall validate any processes for production and service provision where the resulting output cannot be or is not verified by subsequent monitoring or measurement and as a consequence, deficiencies become apparent only after the product is in use or the service has been delivered.
Validation shall demonstrate the ability of these processes to achieve planned results consistently
The organization shall document procedures for validation of processes, including:
(a) defined criteria for review and approval of the processes.
(b) equipment qualification and qualification of personnel.
(c) use of specific methods, procedures and acceptance criteria.
(d) as appropriate, statistical techniques with rational for sample size.
(e) requirements for records (see 4.2.5).
(f) revalidation,
(g) approval of changes to the processes.
The organization shall document procedures for the validation of the application of computer software used in production and service provision. Such software applications shall be validated prior to initial use and as appropriate, after changes to such software or its application. The specific approach and activities associated with software validation and revalidation shall be proportionate to the risk associated with the use of the software, including the effect on the ability of the product to conform to specifications.
Records of the results and conclusion of validation and necessary actions from a validation shall be maintained (see 4.2.4 and 4.2.5)
ISO 13485: 2016 7.5.7 Particular requirements for validation of processes for sterilization and sterile barrier systems
The organization shall document procedures (see 4.2.4) for the validation of processes for sterilization and sterile barrier systems.
Processes for sterilization and sterile barrier systems shall be validated prior to implementation and following product or process changes, as appropriate.
Records of the results and conclusion of validation and necessary actions from the validation shall be maintained (see 4.2.4 and 4.2.5
FDA CFR 21 Part 820.75 Process validation:
(a) Where the results of a process cannot be fully verified by subsequent inspection and test, the process shall be validated with a high degree of assurance and approved according to established procedures. The validation activities and results, including the date and signatures of the individual(s) approving the validation and where appropriate the major equipment validated, shall be documented.
(1) Each manufacturer shall establish and maintain procedures for monitoring and control of process parameters for validated processes to ensure that the specified requirements continue to be met.
(2) For validated processes, the monitoring and control methods and data, the date performed, and where appropriate, the individual(s) performing the process, or the major equipment used shall be documented
(c) When changes or process deviations occur the manufacturer shall review and evaluate the process and perform revalidation where appropriate. These activities shall be documented.
Summary of regulatory requirements:
So both the ISO 13485:2016 and the FDA requirements for process validation are clear on the overall regulatory requirements but provide little instruction on how these validations should be conducted. They do though refer to a document from GHTF (now IMDRF) that is a good overall guide on process validation. click here for document
One more area to clarify is the difference between Validation and Verification and according to the FDA they define them as follows:
Validation: Means confirmation by examination and provision of objective evidence that the particular requirements for specific intended use can be consistently fulfilled.
Verification: Means confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.
Process Validation and Design Validation:
Design validation is conducted for the device itself and is carried out to proof, and produce evidence, that it meets user needs, specifications, and the intended use. Process validation focuses on the production processes for the device. For fast-tracking your design implementation the design and process validations can overlap to some degree as the design reaches its final stages. However timing is important as any changes made to the design will likely require process revalidation.
Steps in the Process Validation plan:
The very first step is determination if process validation is required for each manufacturing process involved in the production of your medical device. The process validation decision tree below is referenced in the GHTF Process Validation Guidance:
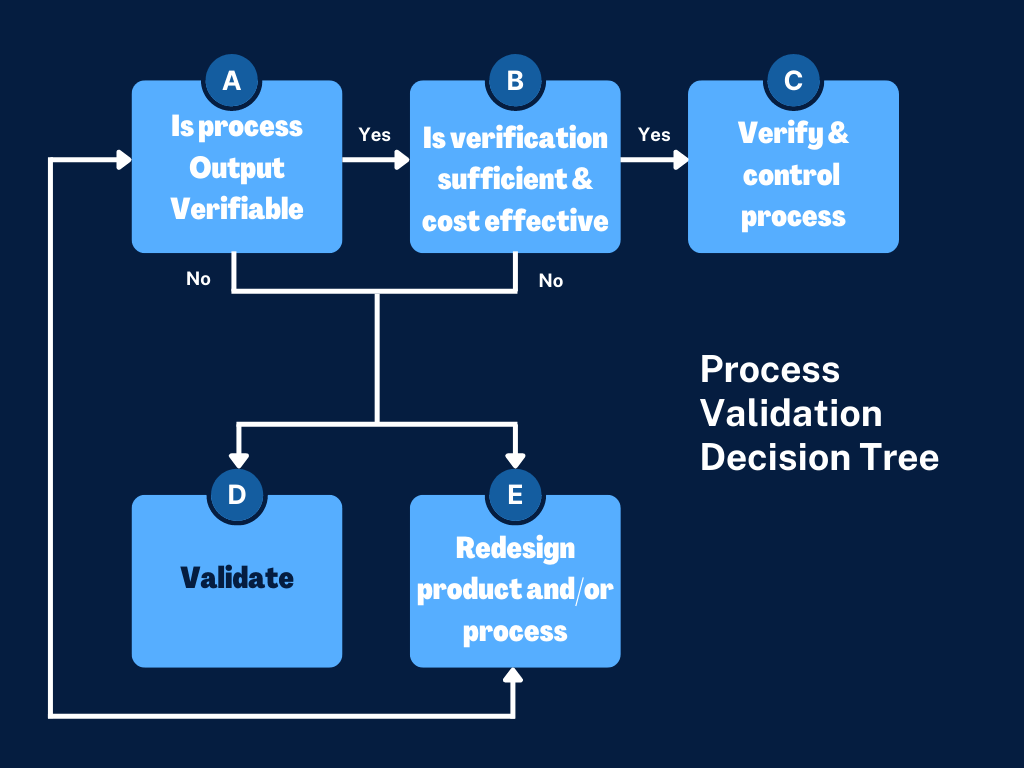
Just to add some background on what production processes should be considered for validation here is some general guidance from GHTF and some other common examples:
- Sterilization and sterile packaging sealing
- Clean room ambient conditions
- Heat treating, welding, soldering, plating, etc.
- Cleaning product in process and finished product
- Bar coding equipment and process
So if its determined that validation is required, the next step is to form a validation team and start the validation process steps as outlined in the diagram below:
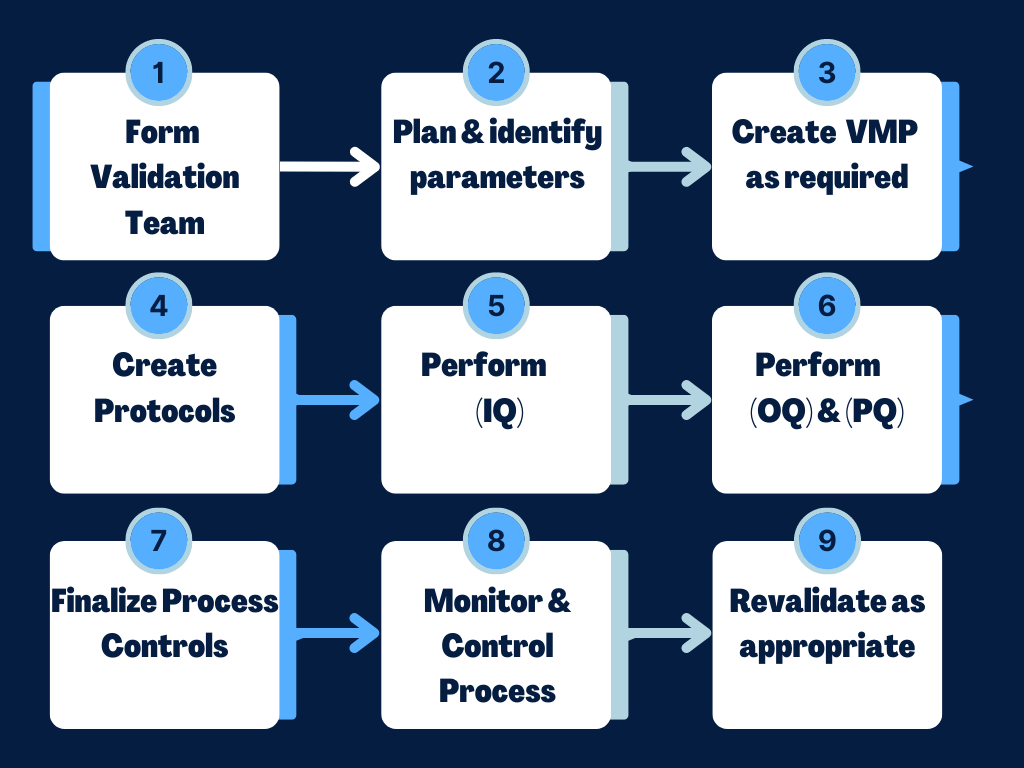
Target validation conclusion:
By the end of the IQ, OQ and PQ validations the following should be answered and determined if the process is stable and capable.

??????????Form Validation Team:
In most cases, other than the simplest validations, a cross-functional team should be formed to plan and oversee the validation activities. Just a very general guideline, in my experience the magic number for team efficiency is trying to not go over 5 to 7 team members. Its ok to bring in other area experts occasionally as required, e.g. regulatory, microbiologist, marketing, etc.
Members of the validation team will depend on the process being validated but can include personnel with expertise in, and representing:
- Quality Assurance
- Engineering
- Manufacturing
- Research & Development
- Inspection and Test
See our blog post on the importance of early training by clicking here
?????????????Team members need to be qualified in the areas they represent, and who know the product and process very well. They also need to be aware of defects and errors that may be encountered as part of their area of responsibility and specific to the product being validated.
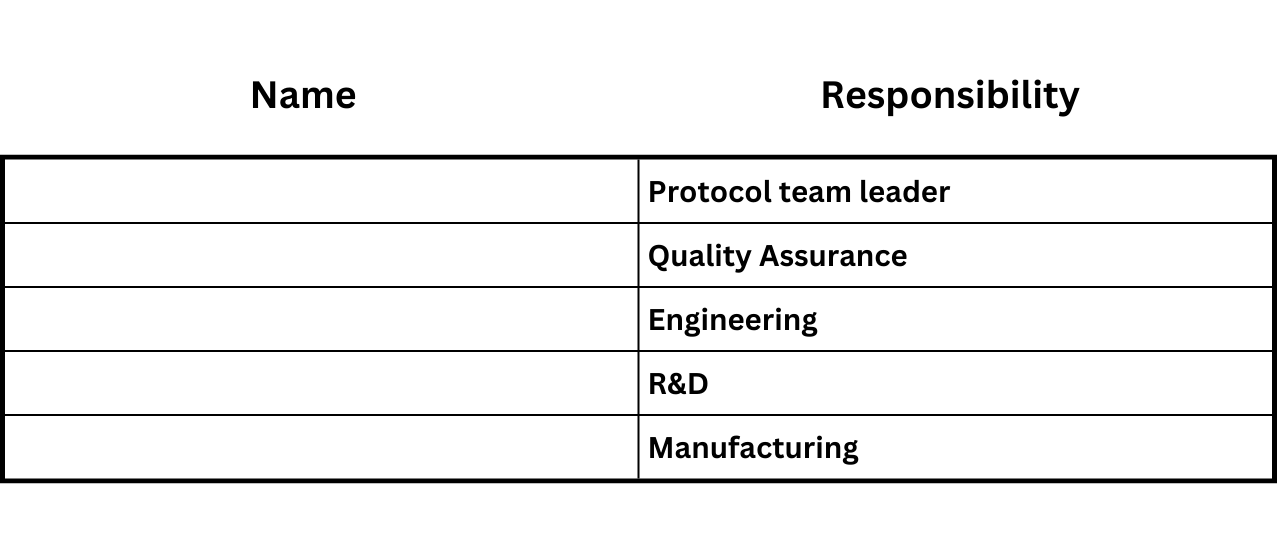
The team leader needs to be trained in the process validation procedure and other team members be familiar with the overall validation process. Each of the validation team members will be listed in the protocols along with the areas they represent. This may appear similar to below:
Team members and Responsibility Matrix:
(List validation team members and area of responsibility for running the protocol)
Once the team has been formed, the next step is to plan the approach and define the requirements. The overall approach may require a Master Validation Plan, Factory Acceptance Test?s and Software Validation. These are described briefly next, before getting into the details of writing protocols and conducting IQ, OQ and PQ
Master Validation Plan, Factory Acceptance Test, and Software Validation:
Master Validation Plan (VMP):
Not a requirement, but is recommended by GHTF and in my experience adds value to major validation projects and should be considered for more complex validations, new plant validation or significant expansion, multiple processes to be validated etc. The Master Validation Plan should include:
- Define the product and process flow as well as the relationship between processes
- Scope of the VMP and identify what needs to be validated and key acceptance criteria, including as appropriate, equipment, processes, buildings, utilities, etc.
- The schedule and sequence, as appropriate, for the validations and resources required.
- Document in a protocol the required individual process validations required.
Factory Acceptance Test (FAT):
FAT is testing conducted at the equipment suppliers factory before delivery to the purchasers site. Not a regulatory requirement for ISO 13485 or the FDA but in many cases for a start- up company, or an established business, that is purchasing new equipment for use in its manufacturing process it makes sense to conduct an FAT where appropriate. This will help provide confidence that the equipment meets specifications and its intended purpose.
A Site Acceptance Test (SAT) can be conducted after the FAT has taken place and the equipment has been delivered and installed. The FAT and SAT can determine whether the new equipment will function as needed and should be included in your Master Validation Plan. It is far better to find any issues before you start the formal validation process and starting with the installation qualification.
Software Validation:
Any software used in the production of your medical device requires validation and is a requirement of both ISO 13485 and the FDA. Also any software used for monitoring, measurement, or as an integral part of your QMS needs to be validated prior to use.
Software validation is another important area and will be covered in detail in a future article.
Writing Protocols and Final Reports
The following are guidelines for completing the opening protocol and the protocol final report:
More detail on protocol content can be found in the GHTF process validation guidance document. click here for document

Also see our blog on priority procedures which includes Process Validation. Click here
1.0 Purpose: Brief description of the purpose of the validation, including the process(s) to be validated, the manufacturing equipment and the products affected.
2.0 Scope: Outline of the scope of the validation, i.e. quantities of the product to be run, number of lots, time period etc.
3.0 Reference documents & attachments: List all of the SOP?s and other documents that will be used during the validation, plus any additional information that may be documented in protocol attachments.
4.0 Validation team members and responsibility matrix: List the validation team members and the areas of responsibility involved in the validation protocol. Ensure each team member is qualified to represent their area and complete any required training before conducting the validation.
5.0 Equipment required: List the manufacturing equipment to be used in the validation including the machine I.D numbers. Ensure any required I.Q?s are completed before running OQ?s.
6.0 Training: Detail any required training required for team members and operators, inspectors and testing personnel before conducting the validation. Ensure appropriate formal training records are documented.
7.0 Flow diagram and description of the process: Include as appropriate flow diagrams of major steps in the process being validated and a description of the process. This may reference an existing or draft SOP.
8.0 Materials required: List production materials used in the validation including part numbers.
9.0 Inspections/Testing to be performed and acceptance criteria: List all quality inspections and testing to be performed with rationale for sample size as well as acceptance criteria. Statistical methods to be used such as sampling, design of experiments, and other statistical techniques can be listed here.
10.0 Procedure: Describe the procedure to be used in running the validation.
11.0 Opening Protocol Approvals: Obtain the required approvals before starting the protocol. An opening protocol meeting may be held with involved departments to communicate the validation plan and answer any issues.
12.0 Results: Document a summary of the results against the acceptance criteria and for each acceptance criteria a pass/fail conclusion. Detailed inspection/test reports may be referenced as attachments to the protocol final report.
Process and product data should be analyzed to determine what the normal range of variation is for the process output. Understanding the normal variation of the output is key in determining whether a process is operating in a state of control and is capable of consistently producing the specified output.
One of the outputs of OQ and PQ is the development of attributes for continuous monitoring and maintenance. Process and product data should also be analyzed to identify any variation due to controllable causes. Depending on the nature of the process and its sensitivity, controllable causes of variation may include:
- Temperature and humidity
- Variations in power supply
- Vibration
- Environmental contaminants
- Purity of process water
- Light
- Human factors (training, shifts, ergonomic, etc.)
- Variability of materials
- Maintenance and wear and tear of equipment
During the running of the protocol any discrepancies or deviations from the documented process plan should be addressed and deviations documented and approved. Each deviation should be evaluated, and a conclusion drawn as to the acceptance or rejection of the results. Process control procedures may require modification and those modifications should be validated as part of the overall process.
13.0 Conclusion: Conclusion of the validation (pass/fail) and any required next steps, i.e. revalidation, inspection plans etc.
14.0 Final Report Approvals: Obtain final required protocol approvals. A closing meeting of appropriate affected departments may be carried out to communicate the validation and results.
Now let?s look at more detail on IQ, OQ, and PQ:
Installation Qualification (IQ):
- Simply put, IQ means is it installed correctly? Important IQ considerations to include in the IQ protocol are the checking of:
- Equipment design features (i.e. materials of construction cleanability, etc.)
- Installation conditions (wiring, utilities, functionality, etc.)
- Calibration, preventative maintenance, cleaning schedules
- Safety features
- Supplier documentation, prints, drawings and manuals
- Software documentation and testing
- Spare parts list and ordering of required parts to support initial production
- Environmental conditions (such as clean room requirements, temperature, humidity)
Copies of the suppliers? qualification studies should be used as guides, to obtain basic data, and to supplement installation qualification. However, it is usually insufficient to rely solely upon the validation results of the equipment supplier. Each medical device manufacturer is ultimately responsible for evaluating, challenging, and testing the equipment and deciding whether the equipment is suitable for use in the manufacture of a specific device(s). The evaluations may result in changes to the equipment or process.
- US FDA requirements under CFR 820.70 Production and Process Controls, section (g) is outlined as follows and should be considered in your IQ:
- Equipment: Each manufacturer shall ensure that all equipment used in the manufacturing process meets specified requirements and is appropriately designed, constructed, placed, and installed to facilitate maintenance, adjustment cleaning, and use.
- Maintenance schedule: Each manufacturer shall establish and maintain schedules for the adjustment, cleaning, and other maintenance of equipment to ensure that manufacturing specifications are met. Maintenance activities, including the date and individual(s) performing the maintenance activities shall be documented.
- Inspection: Each manufacturer shall conduct periodic inspections in accordance with established procedures to ensure adherence to applicable equipment maintenance schedules. The inspections, including the date and individual(s) conducting the inspections, shall be documented.
- Adjustment: Each manufacturer shall ensure that any inherent limitations or allowable tolerances are visibly posted on or near the equipment requiring periodic adjustments or are readily available to personnel performing these adjustments.
Automated processes: Each manufacturer shall validate computers or data processing systems that are used as part of production processes.
Operational Qualification (OQ):
Once the IQ is completed and results reported and approved the OQ can proceed.
In the OQ the the process parameters are challenged to assure that they will result in a product that meets all defined requirements under all anticipated conditions of manufacturing, i.e., worst case testing.
During routine production and process control, it is desirable to measure process parameters and/or product characteristics to allow for the adjustment of the manufacturing process at various action level(s) and maintain a state of control. These action levels should be established and documented during the OQ process validation to determine the robustness of the process and ability to avoid approaching ?worst case conditions.?
Running the OQ should include running at anticipated minimum, maximum and mid control limits.
Typically at the start of the OQ and before you start running product you will monitor the process until it becomes stable and runs at standard operating conditions. Adjustments may need to be made until this condition is achieved and this will become a standard part of your operating instructions for normal production. Once the process is stable and operating under specified conditions you can start running product for the OQ.
Depending on the process being validated the OQ should consider including the following:
- Process control limits (time, temperature, pressure, line speed, setup conditions, etc.)
- Software parameters
- Raw material specifications
- Process operating procedures
- Material handling requirements
- Process change control
- Training
- Short term stability and capability of the process, (control charts)
- Use of statistically valid techniques to establish key process parameters and statistically designed experiments to optimize the process
Once the QQ is complete and the results have been documented and approved then the PQ may proceed.
Performance Qualification (PQ):
In this phase the key objective is to demonstrate the process will consistently produce acceptable product under normal operating conditions. PQ considerations include:
- Actual product and process parameters and procedures established in OQ
- Acceptability of the product
- Assurance of process capability as established in OQ
- Process repeatability, long term process stability
Challenges to the process should simulate conditions that will be encountered during actual manufacturing. Challenges should include the range of conditions as defined by the various action levels allowed in written standard operating procedures as established in the OQ phase. The guideline for running validations used to be running three production lots or batches, but is now changed to; the challenges should be repeated enough times to assure that the results are meaningful and consistent.
Process and product data should be analyzed to determine what the normal range of variation is for the process output. Knowing the normal variation of the output is crucial in determining whether a process is operating in a state of control and is capable of consistently producing the specified output.
One of the outputs of OQ and PQ is the development of attributes for continuous monitoring and maintenance. Process and product data should also be analyzed to identify any variation due to controllable causes. Depending on the nature of the process and its sensitivity, controllable causes of variation may include:
- Temperature
- Humidity
- Variations in electrical supply
- Vibration
- Environmental contaminants
- Purity of process water
- Light
- Human factors (training, ergonomic factors, stress, etc.)
- Variability of materials
- Wear and tear of equipment
Appropriate measures should be taken to eliminate controllable causes of variation. Eliminating controllable causes of variation will reduce variation in the process output and result in a higher degree of assurance that the output will consistently meet specifications.
????
Revalidation:
It?s a reality that revalidation will be required at some point, and pretty much guaranteed for a start-up medical device business that has just developed its product design and manufacturing processes. As it moves into production and distribution, improvements are likely to be identified.
Any significant design or process changes needs to follow appropriate procedures including, Control of Design & Development Changes, Risk Management, and Process Validation.
A partial or complete revalidation should be considered for any significant changes including to software/hardware/process modifications, equipment relocation, product design, specification change, and raw material changes.
Revalidation may also be required for any significant quality issues or trends identified by the ongoing measuring, monitoring and analysis procedures. The corrective and preventive action process can also trigger the need for revalidation.
Another factor in determining the need for revalidation is time period and risk. Periodic revalidation is not a requirement, but you may decide that specific processes should be considered for revalidation every 2 or 3 years even if the process is running without issues. Examples of this could be sterilization or sterile sealed packaging. The timeframe should be defined and documented.
A complete IQ, OQ and PQ may not be required in the revalidation and may only require a PQ to confirm all requirements continue to be meet. A rational for this decision should be documented in the protocol.
How can Fast-Track QMS Consultants help:
Want to learn more about Fast-Track Consulting and the document bundles and consulting services we can provide including a proven Process Validation Procedure and Protocol Template
We have experienced experts in process validation in many areas including, facility validations, equipment installation validations, manufacturing process validations, packaging, sterilization, and measuring/testing equipment validations.
????????????????????
Also we can assist with the design of your master validation plans and individual protocols as part of our consulting services.