

In my 25+ years of experience leading quality management systems I have become convinced that the CAPA (Corrective and Preventive Action) procedure and following it with passion can be one of the most important and value-added processes in your quality management system.
It can be a continuous improvement process resolving customer complaints and feedback issues, reducing internal nonconformance?s which can result in significant cost savings, resolving internal and external audit findings, and also of course improving product quality, customer satisfaction and safety of the medical device.
As an integral part of the quality systems ongoing monitoring, measurement and analysis process, CAPA?s can be used to identify and resolve quality problems, reverse negative quality trends and also play a role in keeping quality objectives on track.
That?s a lot of benefits right, and if your still not convinced, then consider your corrective and preventive action procedure will for sure always be audited by external auditors including the FDA and ISO 13485:2016 certification bodies. In fact the FDA specifically for a level 1 QST inspection, sometimes referred to as a ?CAPA Plus One? will start with a focus on CAPA?s including customer complaints, before moving onto one other of the 4 subsystems.
In one of my most memorable FDA QSIT inspections, we were expecting to be audited on a handful of our CAPA?s during the audit, but the inspector asked to see 30 CAPA?s - and right from the start! We then spent the next 3 days going through each in detail. Another good reason for you to have a robust CAPA procedure and to follow it thoroughly.
The bottom line is that a good CAPA process can improve device quality and safety, drive improved customer satisfaction, reduce costs, and result in successful compliance audits.
Now that your convinced, let?s get into the detailed requirements for CAPA for ISO 13485 and the FDA.
The CAPA requirements to meet ISO 13485:2016 Section 8.5 and FDA CFR 21 Part 820.100
ISO 13485:2016:
Section 8.5, 8.5.1, 8.5.2 and 8.5.3 states the following:
The organization shall identify and implement any changes necessary to ensure and maintain the continued suitability, adequacy and effectiveness of the quality management system as well as medical device safety and performance through the use of the quality policy, quality objectives, audit results, post-market surveillance, analysis of data, corrective actions, preventive actions and management review.
8.5.2 Corrective action
The organization shall take action to eliminate the cause of nonconformance in order to prevent recurrence. Any necessary corrective actions shall be taken without undue delay.
Corrective actions shall be proportionate to the effects of the nonconformities encountered.
The organization shall document a procedure to define requirements for:
- (a) reviewing nonconformities (including complaints)
- (b) determining the causes of nonconformities
- (c) evaluating the need for action to ensure that nonconformities do not recur
- (d) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation.
- (e) verifying that the corrective action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device
- (f) reviewing the effectiveness of corrective action taken.
Records of the results of any investigation and of action taken shall be maintained

Corrective Actions:
So it?s very clear on the requirements for the corrective action procedure from ISO 13485 and the same is true for preventive actions as follows as well as the FDA requirements.
ISO 13485:2016:
8.5.3 Preventive action
The organization shall determine action to eliminate the causes of potential nonconformities in order to prevent their occurrence. Preventive actions shall be proportionate to the effects of the potential problems.
The organization shall document a procedure to describe requirements for:
- (a) determining potential nonconformities and their causes
- (b) evaluating the need for action to prevent occurrence of nonconformities
- (c) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation.
- (d) Verifying that the action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device
- (e) Reviewing the effectiveness of the preventive action taken, as appropriate.
Records of the results of any investigation and of action taken shall be maintained.
FDA CFR 21 Part 820.100 Corrective and preventive action:
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product and other sources of quality data to identify existing and potential causes of nonconforming product, or other quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems.
(2) Investigating the cause of nonconformities relating to product, process, and the quality system.
(3) Identifying the action(s) needed to correct and prevent reoccurrence of nonconforming product and other quality problems.
(4) Verifying or validating the corrective and preventive action to ensure that each action is effective and does not adversely affect the finished device.
(5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems.
(6) Ensuring that information related to quality problems or nonconforming product is disseminated to those responsible for assuring the quality of such product or the prevention of such problems and,
(7) Submitting relevant information on identified problems quality problems, as well as corrective and preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.
Despite the clear regulatory requirements for CAPA, many medical device companies fail to have robust procedures in place, or they have significant gaps. In my experience, the most common issues fall into the following categories:
1. No documented CAPA procedure.
2. Failure to verify or validate the effectiveness of the CAPA.
3. Procedures for CAPA not adequately established.
4. CAPA process almost totally managed and carried out by the quality department and lack of cross-functionality.
5. Every nonconformance turned into a CAPA leading to burdensome process with priorities not based on risk and priorities.
6. Poor or no root cause analysis
7. Inadequate effectiveness checks.
So here are a few key actions any medical device quality system and CAPA procedure should include to avoid inspection findings from the FDA or ISO auditors and to provide the most effective CAPA procedure to help your medical device company be successful.
Key actions for your CAPA:
1) Well defined CAPA Procedure
2) Cross functional participation in the CAPA process
3) Prioritize the CAPA?s
4) Follow each CAPA process step
1. Well defined CAPA Procedure:
Having a well-defined CAPA procedure which includes the requirements from FDA CFR 820 and ISO 13485:2016 is obviously an issue based on the items 1 through 7 listed above. Even for a start-up medical device company who may not have the experienced quality team resources the requirements in the standards are clear and specific. What may not be so apparent is the understanding of why each defined requirement is so important and the effort required for each CAPA step, in order to meet the intent and effectiveness of the CAPA procedure and the regulatory requirements.
One solution is to take a look at what we offer at Fast-Track QMS Consultants and contact us for information on our CAPA procedure, training on CAPA, plus consulting as required. We do have the experience and can help you avoid some of the mistakes we have encountered.
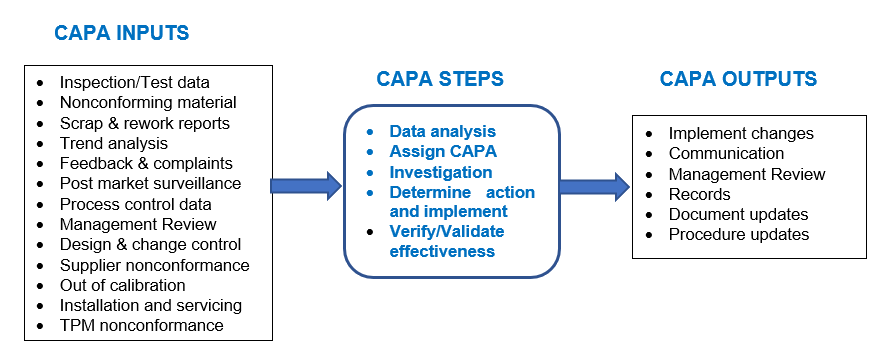
A good CAPA procedure and process starts with monitoring, measuring and analyzing the inputs for CAPA as shown in the diagram below. Once the analysis shows a CAPA is required then the CAPA is assigned to the responsible department and individual leader to form a cross functional team as appropriate, to complete the investigation. Actions are then determined, implemented and verified for effectiveness before closing the CAPA. Each step is reviewed and approved according to the CAPA procedure.
2. Cross functional participation in CAPA process:
The CAPA procedure and management of the process should be managed by the Quality function, and this is frequently the case for medical device companies. The Quality leader normally makes the decision on when a CAPA is needed and approves the issuance of the CAPA to the appropriate person. That makes sense, however best practice is to involve other key functions in your CAPA process to make it a truly cross functional effort and responsibility and to ensure CAPA?s are a priority for the whole company and not just for the Quality department.
The most effective practice I have been a part of and highly recommend is the use of a cross functional CAPA Review Board (CRB), or some companies use a Management Review Board (MRB). Ideally, this board would include leaders from quality, regulatory, engineering and manufacturing operations, as well as specialists as required.
CRB members will need to be trained in the CAPA procedure as of course will all those assigned to work on individual CAPA?s. Make sure you have training records documented. The CRB should meet regularly and at a minimum on a monthly basis.
Here is a sample CRB meeting agenda:
1. Any follow up items from the previous meeting.
2. Review open CAPA?s and determine if any follow up action required.
3. Review major quality indicators, i.e. customer complaints, nonconforming product, audits, etc., to determine if any additional CAPA?s may be required.
4. Invite to present at the meeting any leaders of CAPA?s that may be behind the target completion dates or otherwise running into challenges. This can be very effective in keeping CAPA?s on track and major benefit of a cross functional ownership.
5. Review CAPA metrics against target objectives including:
- Number of open CAPA?s
- Average time to close CAPA?s
- Target CAPA closure time
- Trending of CAPA process
- Longest open CAPA
- Any repeat CAPA?s
6. Issue minutes of the meeting with any decisions made or follow up actions required.
3. Prioritize the CAPA?s:
I have seen overuse of CAPA?s where priority issues were lost in the weeds, and underuse, where CAPA?s were not being issued for repeat or significant quality issues.
Regulatory authorities do not expect you to issue a CAPA for every customer complaint or for every nonconformance, nor is it an efficient use of resources. Not every CAPA you issue will have the same priority so base your priority on risk, and make sure resources are focused on those with higher risk first.
It is important that you issue CAPA?s for systematic issues, or to resolve negative quality trends and to prioritize those CAPA?s so to focus resources on resolving the issues in a timely manner.
Also as ISO 13485: 2016 8.5.2 states; Corrective actions shall be proportionate to the effects of the nonconformities encountered.
4. Follow each CAPA process step:
This is obvious right? Not so, and unfortunately, I have seen many instances where one of the CAPA process steps was inadequately followed or was missed all together. Examples have included:
- not documenting the action that was taken to contain or correct the issue,
- lack of detail on the investigation and root cause analysis
- parts of the CAPA plan not completed
- no or little description of the corrective/preventive actions taken
- risk analysis not considered or carried out as appropriate
- review and approval of each CAPA process step not completed
- no documented effectiveness plan and results
- CAPA completion target dates missed with no explanation or approvals from the CRB
So don?t take short cuts with the CAPA procedure or process, and to repeat,
CAPA is one of, if not the most important quality system elements that a medical device company can have.
Using the CAPA procedure can help your ISO 13485:2016 Certification and needs to be used throughout your QMS Implementation. Discover more ...
To learn more about Fast-Track QMS Consulting and the products and consulting services we can provide, including a CAPA SOP and forms then just click here