

I still recall early in my career, when I was formally introduced to, and required to perform by a corporate QMS procedure, a risk analysis of the design I was working on. This was before moving into the quality management world, and I was heading up a design and development team at the time and I remember my initial skepticism. My attitude was, ?hey I know the risks, why do I need to conduct this formal documented risk analysis?, I have already considered the risks for the use of this product by the customer.
The good news is I learned very quickly as a result of this very first risk analysis the purpose, intent and true benefits of using a formal Risk Management process
This first experience included completing with a cross functional team a DFMEA. To our surprise we found a risk involving potential for a major product failure due to component fatigue, that no one on the team had thought of, and as the design was unique there was no history to guide us. We ran a test and sure enough it failed. The result was that this risk analysis enabled us to modify the design so that there was no failure during validation and no customer harm. Convinced me of the importance of risk management.
In this article we will look briefly at each of the risk related requirements in ISO 13485:2016, elements 4, 6, 7 and 8, and then review the details of the risk management process as required by ISO 14971:2019
Risk mentioned in 10 clauses of ISO 13485:2016
Introduction
A good place to start is with the definition of risk and risk management from ISO 13485:2016 Section 3 Terms and Definitions:
(Source ISO 14971)
Risk: Combination of the probability of occurrence of harm and the severity of that harm
Risk Management: Systematic application of management policies, procedures and practices to the tasks of analysing, evaluating, controlling and monitoring risk.
When a patient requires surgery or any treatment using a medical device, they are not going to think about the quality or risks involved with the medical device, and they place their trust with the surgeons and medical staff.
Risk Management and Quality is the responsibility of the device manufacturer who designs, manufactures and markets the device.
That is why risk management is such a critical part of the medical device world and the quality management system. It should never be just another ?check-the-box? activity and needs to have full support of management and be a priority within the QMS. Top management and team leaders, need to be involved, and starting from ensuring there are adequate, trained resources to conduct risk management and to raise the awareness of its importance.
One best practice I have seen from executive management was with the first medical device company I worked with,
and which had a lasting impression on how I thought about medical device quality and the risk management required.
This was where the executive management would invite surgeons from local hospitals to give a talk to all employees
on the importance of their product quality.
Part of that presentation was the doctors asking employees to just imagine it was members of their families, or
themselves, being treated with the use of the medical device they had helped to design and build.
As you might imagine that was very effective and something that has stayed with me throughout my medical device
quality management career. Perhaps you can also keep this in mind as you read this guide to risk management.
Risk requirements to meet ISO 13485:2016
The following is a summary of the requirements for a risk-based approach that are set out in ISO 13485:2016:
(For complete requirements refer to the ISO 13485:2016 Standard)
Section 4 Quality Management System
- Under general requirements 4.1.2; It states the organization shall apply a risk based approach to the control of the appropriate processes for the quality management system.
Note: This requirement is for a risk-based approach for the control of appropriate processes and depending on the
medical device can go beyond those specifically called for in the other sections of the Standard.
- For outsourced processes 4.1.5; When the organization selects to outsource any processes that affects product conformity to requirements, the controls shall be proportionate to the risk involved and the ability of the supplier to meet requirements.
- 4.1.6; Requires validation of the application of computer software used in the quality management system prior to use. The specific approach and validation activities shall be proportionate to the risks associated with the use of the software.
Section 6 Resource Management
- Under 6.2; checking the effectiveness of training for personnel performing work affecting product quality, the methodology used to check effectiveness to be proportionate to the risk associated with the work.
Section 7 Product Realization
- 7.1; Requirement to document one or more processes for risk management for product realization and to maintain records.
- Design and development inputs 7.3.3; To be determined and records maintained to include applicable output(s) of risk management.
- Control of design and development changes 7.3.9; shall include evaluation of the effect on the inputs or outputs of risk management.
- 7.4.1; Purchasing process and the criteria for the evaluation and selection of suppliers shall be proportionate to the risk associated with the medical device. Under 7.4.3 Verification of purchased product the inspection to be based on the supplier evaluation and proportionate to the risk associated with the purchased product.
- 7.5.6; Validation of the application of computer software used in production and service prior to use and to be proportionate to the risk associated with the use of the software, including the effect on the ability of the product to conform to specifications.
- 7.6; Validation of the application of computer software used for monitoring and measuring of requirements. The specific approach and activities associated with software validation and revalidation shall be proportionate to the risk associated with the use of the software.
Section 8 Measurement, Analysis and Improvement
- 8.2.1; Feedback requires that information gathered in the feedback process shall serve as potential input into risk management for monitoring and maintaining the product requirements as well as the product realization or improvement processes.
ISO 14971 Risk Management
ISO 14971:2019 Medical devices ? Application of risk management to medical devices, is the Standard for risk management referenced in the ISO 13485:2016 Standard.
This document should be included in your controlled external documents and is an excellent standard for explaining the requirements, expectations, and the stages of a risk management process for medical device companies.
It includes terminology, principles and a process for risk management, including software as a medical device and in vitro diagnostic medical devices.
Another useful document is ISO/TR 24971:2020 Medical devices ? Guidance on the application of ISO 14971. This document provides guidance on the development, implementation and maintenance of a risk management system for medical devices according to ISO 14971:2019.
Definitions
It is important to understand definitions under Risk Management as defined in ISO 14971 as the terminology can sometimes be used incorrectly, i.e. Risk Management and Risk Analysis Use correct definitions consistently as shown below to ensure clear communication.
???????????
Risk Management: Systematic application of management policies, procedures, and practices to the tasks of analyzing,
evaluating, controlling and monitoring risk.
Risk: Combination of the probability of occurrence of harm and the severity of that harm.
Hazard: Potential source of harm.
Hazardous Situation: Circumstances in which people, property, or the environment are exposed to one or more
hazards.
Harm: Injury or damage to the health of people or damage to property or the environment.
Severity: Measure of the possible consequences of a hazard.
Risk Analysis: Systematic use of available information to identify hazards and to estimate the risk.
Risk Estimation: Process used to assign values to the probability of occurrence of harm and the severity of that harm.
Risk Evaluation: Process of comparing the estimated risk against given risk criteria to determine the acceptability of
the risk.
Risk Assessment: Overall process comprising a risk analysis and a risk evaluation.
Risk Control: Process in which decisions are made and measures implemented by which risks are reduced, or
maintained within, specified levels.
Risk Management Plan: A product level document that identifies the risk management activities anticipated and
planned throughout the products life cycle.
Risk Management File: File to keep risk management activities, documentation, and records.
Residual Risk: Risk remaining after risk control measures have been taken
Risk Management Process
For medical device design, development and manufacturing it is essential to have a defined and documented Risk Management process fully implemented. It must have support of top management, and those involved should include a cross-functional engagement in the risk management process.
Also it is best practice to have at a minimum the risk management process leader for each project formally trained in the whole risk management process, and all participants trained in the requirement of the companies procedure.
The Risk Management process includes:
- Risk Management Planning
- Risk Analysis
- Risk Evaluation
- Risk Controls
- Overall Residual Risk Acceptability
- Risk Management Review
- Production and Post-Production information and review
Risk Management Plan
The Risk Management Plan is a product level document, and the need for risk identification is determined for new designs, changes to current designs and processes, or can also be on the basis of information and trends regarding the performance and effectiveness of the QMS.
Once the need for Risk Management has been identified and depending on the magnitude and scope of the project or need, a Risk Management Plan should be initiated.
This plan should identify the Risk Management activities anticipated and planned throughout the project?s life cycle. The Risk Management plan is dynamic and should be reviewed and updated as required.
The Risk Management Plan to include:
??????????
- Scope of the Risk Management activities and definition of the product or process. It is possible to have multiple similar products or processes in a single Risk Management Plan.
- Description of the intended use of the product(s) or process(s).
- Identify all Risk Management activities planned throughout the product/process life cycle.
- Define roles and responsibilities of the Risk Management team that will be participating, reviewing and approving risk documentation.
- Criteria for the product or process risk acceptability.
- Methods to verify Risk Control measures after implementation and reduction of risks to the pre-established acceptable levels.
- How post-production information will be captured and fed into Risk Management activities for the product/process.
- Risk Management Plans should be reviewed and updated throughout the products life cycle.
Risk Management File:
A Risk Management File (RMF) is generated and maintained to keep all of the risk management activities, documentation, and records.
A Risk Management File contains evidence of the following:
- Risk Management Plan
- Risk Analysis
- Risk Evaluation
- Risk Controls
- Evaluation of Overall Risk Acceptability
- Risk Management Review
- Production and Post- Production Risks
Risk Management Files may as an alternative refer to the location of such records, i.e. DHR?s, CAPA?s etc. This can be a challenge if using a paper based approach and control of these documents and records can certainly be one of the advantages of using an eQMS system.
Our website Fast-Track QMS Consultants has contact information for approved partners where you can learn more on this.
Risk Analysis
Risk Analysis and Risk Evaluation are in my experience, normally carried out at the same time and using different techniques including FMEA?s, preliminary hazards analysis and fault tree analysis, as appropriate.
FMEA?s are a reliability tool that assumes single-fault failures as part of the analysis. Risk Management is broader than just failures; risks exist when medical devices are used without failure modes.
Any Risk Analysis conducted must identify the medical device or process, the intended use, the team members involved, scope and date. It is also important to consider off-label hazards as well as those from the intended use.
This is where it can be extremely beneficial to obtain input from functional areas outside of just design and process engineers, and include marketing, sales and end users.
Risk Evaluation
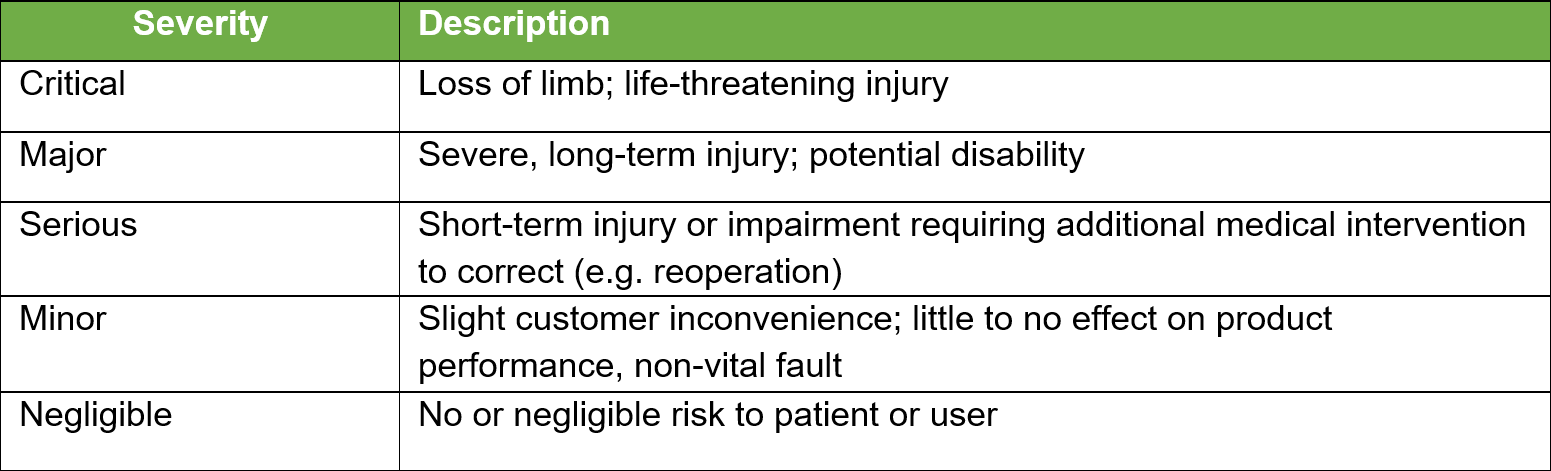
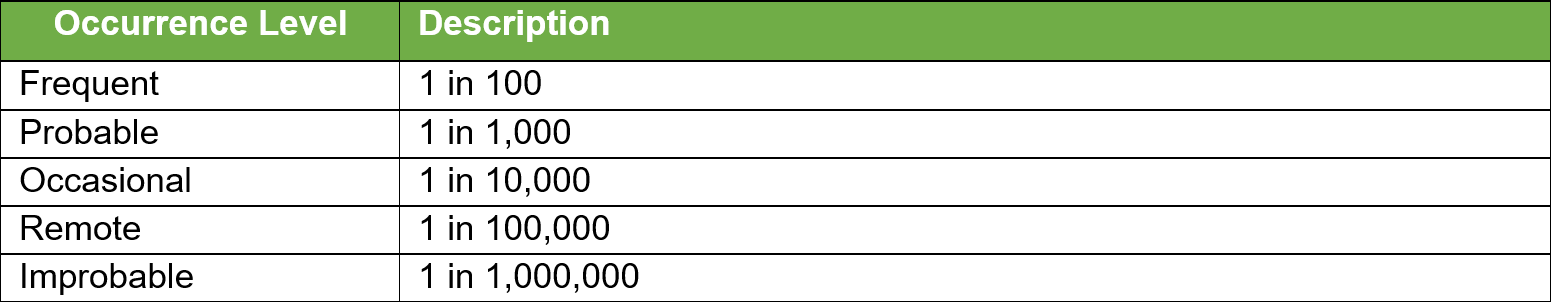
Once all the known and/or anticipated risks have been identified and estimated, these risks need to be evaluated to determine if risk reduction is required. Using the results of the Risk Analysis and with reference to the tables below determine and identify which risk zones are acceptable and which require risk reduction.
For the US Market the low zone is normally acceptable and the high zone unacceptable. The medium zone can fit into ?as low as reasonably practical?. Items in the high-risk zone require risk reduction and those in the medium zone should also be considered for risk reduction. For product sold in the EU the MDR requires reduction of risks as far as possible for all levels.

Risk Controls
Once the Risk Analysis and Risk Evaluation are completed the next step is to identify Risk Controls. Items identified as requiring Risk Reduction are about reducing those identified risks to acceptable levels.
Risk control options should be considered in the following priority order:
- By modifying the design to build in safety and this is always the preferred option
- Protective measures in the actual medical device and/or in the manufacturing process
- Information for safety such as labeling and instructions for use
Where possible it is best practice to include multiple Risk Controls to reduce risk, i.e. by design and information on labeling.
After the risk controls are identified they need to be implemented, verified, and the effectiveness determined. Records of these actions to be documented. Where appropriate also need to evaluate if new risk controls introduce any new hazards.
If the first priority risk control option is used and incorporated into the design, then using the design and development steps of Design Outputs, Design Verification and Design Validation will make verifying the effectiveness of these controls part of your Design Controls process.
Overall Residual Risk Acceptability
In addition to the evaluation of individual risks the overall device risk acceptability is to be evaluated.
If it is determined that the overall residual risk is acceptable the decision is documented in the Risk Management Report along with the rationale.
If you determine that the overall residual risk is not acceptable you may want to go back and determine if every possible measure has been taken to reduce the risk first through Risk Controls. You can also conduct and document a benefit-risk analysis.
Benefit-Risk Analysis
After completing identification of Risk Controls and evaluating residual risks, it maybe that some risks still remain that fall into the unacceptable range. It may then be appropriate to consider conducting a benefit-risk analysis, but only after every possible measure to reduce risks has been taken.
This analysis is to consider if the medical benefits of the medical device outweigh the residual risk. The analysis if conducted must be documented along with objective evidence and rational for why the medical benefits outweigh the unacceptable risks. The key here is medical benefits and never to include financial business factors.
Risk Management Review
On completion of all the steps in the risk management process all activities are to be documented in a Risk Management Report, reviewed and approved, and I would recommend include approval by executive management.
The report should include the plan for evaluating risks in production and post-production. The review plan for post-production would normally be first conducted no later than 6 months after the launch of the product, but should be targeted as appropriate for your medical device.
Production and Post-Production Review
Risk Management is a total product life cycle process, and the Risk Management File is a living document, to be reviewed on a regular documented basis, or as activities and events determine a need for action and update.
Examples where updates to the RMF may be required include:
- Consider and document production related risk management activities and events.
- Ensure post-production processes put in place to support the QMS are feeding into the Risk Management process.
- Feedback and customer complaints tied into where appropriate, the risk management process. Including verification
that any occurrence of harm aligns with what was estimated and any new hazard or hazardous situation is identified.
- Non-conformances and CAPA?s tied into the risk management process.
- Negative quality trends.
Final Thoughts
Risk Management can be a challenging and sometimes difficult process to get embedded and consistently applied, in your quality management system. I highly recommend you give this a high priority and seek out guidance if you need it.
Hopefully this guide helped you with the understanding the fundamentals of Risk Management. If this is a process, you need to implement or will be involved in some way with its application you should use ISO 14971 to make it easier.
Remember the Risk Management process includes:
- Risk Management Planning
- Risk Analysis
- Risk Evaluation
- Risk Controls
- Overall Residual Risk Acceptability
- Risk Management Review
- Production and Post-Production information and review
All to be documented in the Risk Management File
Risk Management needs to be an integral part of Design and Development as well as the other quality management elements listed under the ISO 13485 Risk Requirements section of this article.
Risk Management needs to be active throughout the entire product lifecycle
Risk Management needs to be a priority for start-up medical device companies.
If you would like to learn more from our available eBooks which include application of Risk Management,
the following are 3 that we have available for free download from our website:
Priority QMS Procedures for Start-Up Medical Device Companies
Complete Guide: Medical Device Design and Development
How to Control Design Changes for Your Medical Device
Need training or coaching on Risk Management or any part of your ISO 13485:2016 quality management system? You can check out our Fast-Track QMS Consultants website to learn more on the consulting services and products we offer, and you can also contact us with any questions.
We also have available a proven Risk Management SOP Template including support forms for PFMEA, DFMEA, and Risk Management Plan for purchase and quick download.