

Changes to your medical device design, manufacturing processes or regulatory and standard requirements will inevitably occur and if you?re a start-up company these may be more frequent than with a mature business and device, but in both cases, changes will occur and need to be managed well.
That means you need to have processes and procedures in place to ensure:
- There is a formal controlled process for requesting a change
- Required reviews and approvals of requested changes
- Managing and documenting the change
It?s also a requirement under ISO 13485:2016 and the FDA that design changes to your medical device be documented and controlled. If changes are not carried out appropriately it could lead to major findings or worse, and/or significant product quality and safety issues.
This article will summarize and outline the steps required for a robust change control and change management process that will facilitate the control and documentation of changes effectively as well as meeting the required regulatory and standard requirements.
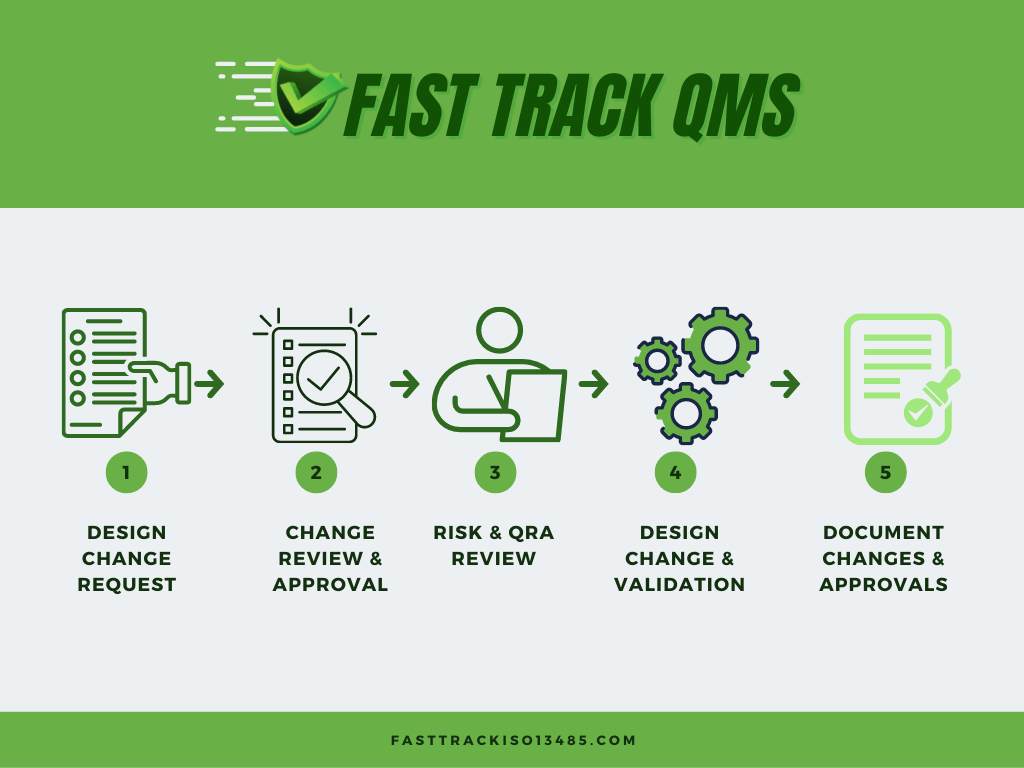
But before we look at each of the process steps shown in above diagram lets take a look at the ISO and FDA requirements:
Control of Design and Development changes to meet ISO 13485:2016 7.3.9 and FDA 21 CFR Part 820.30(i)
ISO 13485:2016: 7.3.9 Control of design and development changes
The organization shall document procedures to control design and development changes. The organization shall determine the significance of the change to function, performance, usability, safety and applicable regulatory requirements for the medical device and its intended use.
Design and development changes shall be identified. Before implementation, the changes shall be:
- a) reviewed.
- b) verified.
- c) validated, as appropriate.
- d) approved.
The review of design and development changes shall include evaluation of the effect of the changes on constituent parts and product in process or already delivered, inputs or outputs of risk management and product realization processes.
????????
Records of changes, their review and any necessary actions shall be maintained. (see 4.2.5)
FDA 21 CFR Part 820.30(i) Design changes
Each manufacturer shall establish and maintain procedures for the identification, documentation, validation or where appropriate verification, review, and approval of design changes before their implementation.
Note: The above outlines the requirements for design changes that occur after the product is in production and that is the focus of this article. However it is also necessary to control changes during the design and development pre-market phase. These changes need to be controlled, validated, reviewed, and documented in your device master record (DMR). This article is focused on changes that are considered minor in nature and that major changes would go through a design and development project as covered in ISO 13485:2016 7.3
Now let?s take a look at each one of the phases for control of design changes for your medical device as shown on the above diagram
Design Change Request:
There are many types of events that can be classified as triggers for design changes and some of the most common ones are:
- Customer complaint negative trend requiring a CAPA and design change.
- Process improvements or changes to manufacturing processes
- Updated regulations or QMS changes
- Label minor changes (not involving addition or change to a claim which may be a new design and development project)
All minor changes should start with a documented design change request, sometimes referred to as an Engineering Change Request (ECR). If you would like to receive a sample of this form, just send us an email at fasttrackiso13485@gmail.com and we will forward you a free copy of our template so you can then use to initiate your change control process.
The Change Request Form should include the following:
- Name of person requesting the change and date requested.
- Control number obtained from the Quality document control team.
- Description of the change
- Reason and justification for the change
- Risk Evaluation including any effect on regulatory requirements
Change Review and Approval:
Once the Change Request is submitted it will need to be reviewed and formally approved and documented on the Change Request Form, by Design Engineering and Quality before the change process can start. This may require a documented design review depending on the complexity of the requested change.
In today?s world there is a better alternative for handling document changes and approvals rather than a manual system. We can recommend one of our partners of Fast-Track QMS Consultants, who offer an easy to use, cloud-based software platform specifically designed to help medical device companies better manage their Design Controls including control of Design Changes.
To learn more, and for a free demo click here: link

Risk and Regulatory Assessment:
A risk assessment should be carried for all but the simplest of changes and completed before the design change process begins. The formal risk assessment can help guide the design as well as its verification and validation plans. The review of design changes along with the risk assessment should include evaluation of the effect of the changes on the product, including product in process and product already distributed.
A regulatory assessment may also be required depending on the nature of the change requested. If it?s a new market for your medical device the regulatory requirements may be different and would need to be considered.
Or if the design change is fairly significant the current market regulatory authority may need to be informed and even in some cases it may be required that you need to obtain the appropriate documented permission from the regulatory body before you can implement the change. This is a critical element due to its potential significant impact on the implementation of the design change.
Risk and Regulatory assessments need to be documented and included with the change control documentation.
Design Change and Validation:
After the change request is approved and the risk assessment is completed including any planned risk reduction actions as appropriate, the design change can proceed.
Design Reviews can be carried out as required throughout the change process.
Verification and Validation of the design change is carried out as appropriate and documented or referenced in the change control file.
Document Changes and Approvals:
All the way through the design change process the decisions you make regarding the change need to be documented. This includes the reason for the change including justification, the risk assessment and any actions taken, details of the change and the verifications and validations carried out.
Once the design change is completed including verification and validation, the appropriate documents, along with the completed Design Change Request (or ECR) and including i.e. specifications, procedures, BOM?s, etc. can be updated, approved and released. This final release is carried out according to your quality management system document control procedures.
How can Fast-Track QMS Consultants help:
Want to learn more about Fast-Track QMS Consulting and the document bundles and consulting services we can provide, including a proven,
?????Control of Design and Development Changes, procedure and supporting templates.
Go to our website at: https://fasttrackiso13485.com/
We have also have procedures for Design and Development, Process Validation, and all other elements of the ISO 13485:2016 Quality System. Our complete Turnkey Document Bundle includes 50 procedures and supporting forms that also cover FDA 21 CFR 820 requirements.
Also we can assist with consultation on any part of your ISO 13485:2016 implementation from planning, through implementation and final certification, or help with any part of your quality management system to suit your needs.