

Before I entered the medical device quality and regulatory world way back in 1998, I had spent time working for a major global automotive company as a QMS leader, and before that as a Design and Development project manager.
It was during those early days that I learned the true benefits of using a formal Design and Development process. As it was automotive there was no FDA or ISO 13485 Design Control regulations involved but the design tools, we used were not that different, i.e. design reviews, risk assessment, documentation, verification and validation, etc.
I still recall when first required by a corporate QMS procedure to conduct design reviews and risk analysis, my initial skepticism. My attitude was, ?hey I know the risks, why do I need to conduct this FMEA?, or ?design review, why do, I have already myself reviewed the design?.
The good news is I learned very quickly the purpose, intent and true benefits of using Design and Development Controls, so by the time I entered the medical device world as a quality and regulatory leader, I had a solid foundation in these requirements, and why they need to be followed, not just to meet regulatory requirements but how they lead to a better and safer product design, that meets the intended use.
In this guide I will explain each step of the design and development process, the controls required by ISO 13485:2016 and FDA, as well as share some of my experience with best practice and how they can benefit your medical device design project and resulting final product design.
We will start with the details of element 7.3 of ISO 13485:2016, as well as a summary of the FDA requirements, and then move into more on each of the design and development steps shown below.
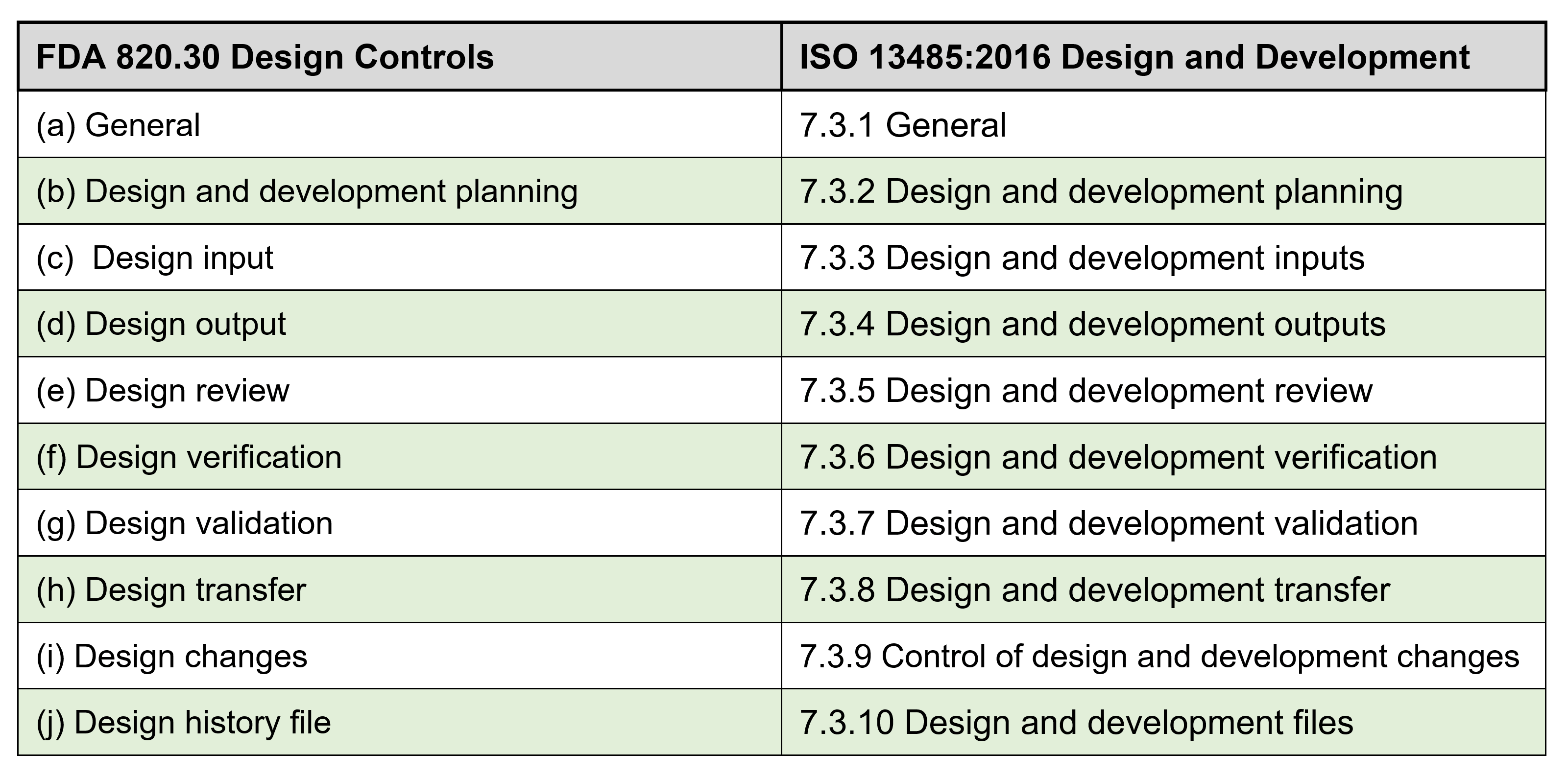
ISO 13485:2016 Requirements for Design and Development
ISO 13485:2016: Sections 7.3.1, through 7.3.10 states the following:
7.3.1 General
The organization shall document procedures for design and development
7.3.2 Design and development planning
The organization shall plan and control the design and development of a product. As appropriate, design and development planning documents shall be maintained and updated as the design and development progresses.
During design and development planning, the organization shall document:
a) the design and development stages;
b) the review(s) needed at each design and development stage;
c) the verification, validation, and design transfer activities that are appropriate at each design and development stage;
d) the responsibilities and authorities for design and development;
e) the methods to ensure traceability of design and development outputs to design and development inputs;
f) the resources needed, including necessary competence of personnel.
7.3.3 Design and development inputs:
Inputs relating to product requirements shall be determined and records maintained. These inputs shall include:
a) functional, performance, usability and safety requirements, according to the intended use;
b) applicable regulatory requirements and standards;
c) applicable output(s) of risk management;
d) as appropriate, information derived from previous similar designs;
e) other requirements essential for design and development of the product and processes.
Those inputs shall be reviewed for adequacy and approved
Requirements shall be complete, unambiguous, able to be verified or validated, and not in conflict with each other.
7.3.4 Design and development outputs:
Design and development outputs shall:
a) meet the input requirements for design and development;
b) provide appropriate information, for purchasing, production and service provision;
c) contain or reference product acceptance criteria;
d) specify the characteristics of the product that are essential for its safe and proper use.
The outputs of design and development shall be in a form suitable for verification against the design inputs and shall be approved prior to release.
Records of the design and development outputs shall be maintained.
7.3.5 Design and development review:
At suitable stages, systematic reviews of design and development shall be performed in accordance with planned and documented arrangements to:
a) evaluate the ability of the results of design and development to meet requirements;
b) identify and propose necessary actions.
Participants in such reviews shall include representatives of functions concerned with the design and development stage being reviewed, as well as other specialist personnel.
Records of the results of the reviews and any necessary actions shall be maintained and
include the identification of the design under review, the participants involved and the date of the review.
7.3.6 Design and development verification:
Design and development verification shall be performed in accordance with planned and
documented arrangements to ensure that the design and development outputs have met the design and development input requirements.
The organization shall document verification plans that include methods, acceptance criteria and as appropriate, statistical techniques with rationale for sample size.
If the intended use requires that the medical device be connected to, or have an interface with, other medical device(s), verification shall include confirmation that the design outputs meet design inputs when so connected or interfaced.
Records of the results and conclusions of the verification and necessary actions shall be maintained.
7.3.7 Design and development validation:
Design and development validation shall be performed in accordance with planned and
documented arrangements to ensure that the resulting product is capable of meeting the requirements for the specified application or intended use.
The organization shall document validation plans that include methods, acceptance criteria and as appropriate, statistical techniques with rational for sample size.
Design validation shall be conducted on representative product. Representee product includes initial production units, batches or their equivalents. The rational for the choice of product used for validation shall be recorded.
As part of design and development validation, the organization shall perform clinical evaluations or performance evaluations of the medical device in accordance with applicable regulatory requirements. A medical device used for clinical evaluation or performance evaluation is not considered to be released for use to the customer.
If the intended use requires that the medical device be connected to, or have an interface with, other medical device(s), validation shall include confirmation that the requirements for the specified application or intended use have been met when so connected or interfaced.
Validation shall be completed prior to release for use of the product to the customer.
????????????
Records of the results and conclusion of validation and necessary actions shall be maintained.
7.3.8 Design and development transfer
The organization shall document procedures for transfer of design and development outputs to manufacturing. These procedures shall ensure that design and development outputs are verified as suitable for manufacturing before becoming final production specifications and that production capability can meet product requirements.
Results and conclusions of the transfer shall be recorded.
7.3.9 Control of design and development changes:
The organization shall document procedures to control design and development changes. The organization shall determine the significance of the change to function, performance, usability, safety and applicable regulatory requirements for the medical device and its intended use.
Design and development changes shall be identified. Before implementation the changes shall be:
a) reviewed
b) verified
c) validated, as appropriate
d) approved
The review of design and development changes shall include evaluation of the effect of the changes on constituent parts and product in process or already delivered, inputs or outputs of risk management and product realization processes.
Records of changes, their review and any necessary actions shall be maintained.
7.3.10 Design and development files:
The organization shall maintain a design and development file for each medical device type or medical device family. This file shall include, or reference records generated to demonstrate conformity to the requirements for design and development and records for design and development changes.
FDA CFR 21 Part 820.30 Requirements ? Summary of differences:
The FDA 21 CFR 820 and the ISO 13485 are very similar, and the table below shows a side-by-side comparison for each of the Design Control elements. There are a couple of minor difference under FDA so if you?re a medical device company that intends to market your device in the US you need meet all the FDA requirements.
A couple of examples that FDA requires that are different than ISO 13485 are:
- 820.30(e) Design Review ?..The procedure shall ensure that participants at each design review include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed.
Note: It?s not a major difference from ISO 13485 for overall requirements, but an independent participant in the reviews needs to be included and make sure you document in the records of the review.
- 820.30(j) Design history file. Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part.
Note: Not really an additional requirement over ISO 13485 under 4.2.3 Medical device file and 7.3.10 Design and development files but can sometimes cause confusion with file name differences. Meet FDA or ISO 13485 and you should be OK.
Introduction:
It is not unusual for start-up medical device companies to already have started designing their device and making early prototypes well before planning the implementation of their ISO 13485:2016 Quality Management System.
This is fine as long as these first prototypes are pure R&D devices and will not be marketed and sold as finished units to customers. Simply put design controls need to start when research ends, and design and development begins, and when you plan to bring your device to market.
Before the formal quality system implementation is started however ,the Design and Development procedure needs to be one of the very first SOP?s to be implemented. This documented procedure needs to used during the design and development of the device design from the design inputs step to the final transfer to production.
Documentation of user needs, design inputs, design process, design outputs, risk assessment, design reviews, design validation and design transfer, are all required parts of the design and development procedure.
For a start-up medical device company it is understood and very common practice that the design and development of the medical device and the full implementation of the quality management system will be taking place in parallel. However there are at least 7 procedures that you should give priority too and have in place before you start your Design and Development project:
1) Design and Development
2) Risk Management
3) Document Controls
4) Records Controls
5) Process Validations
6) Supplier Approval
7) Training
???????????????????????
For advice from Fast-Track QMS Consultants on Priority Procedures for a Start-Up Medical Device Company to Implement First, go to our Guide by clicking on the link below:
Priority QMS Procedures for a Start-Up Medical Device Company
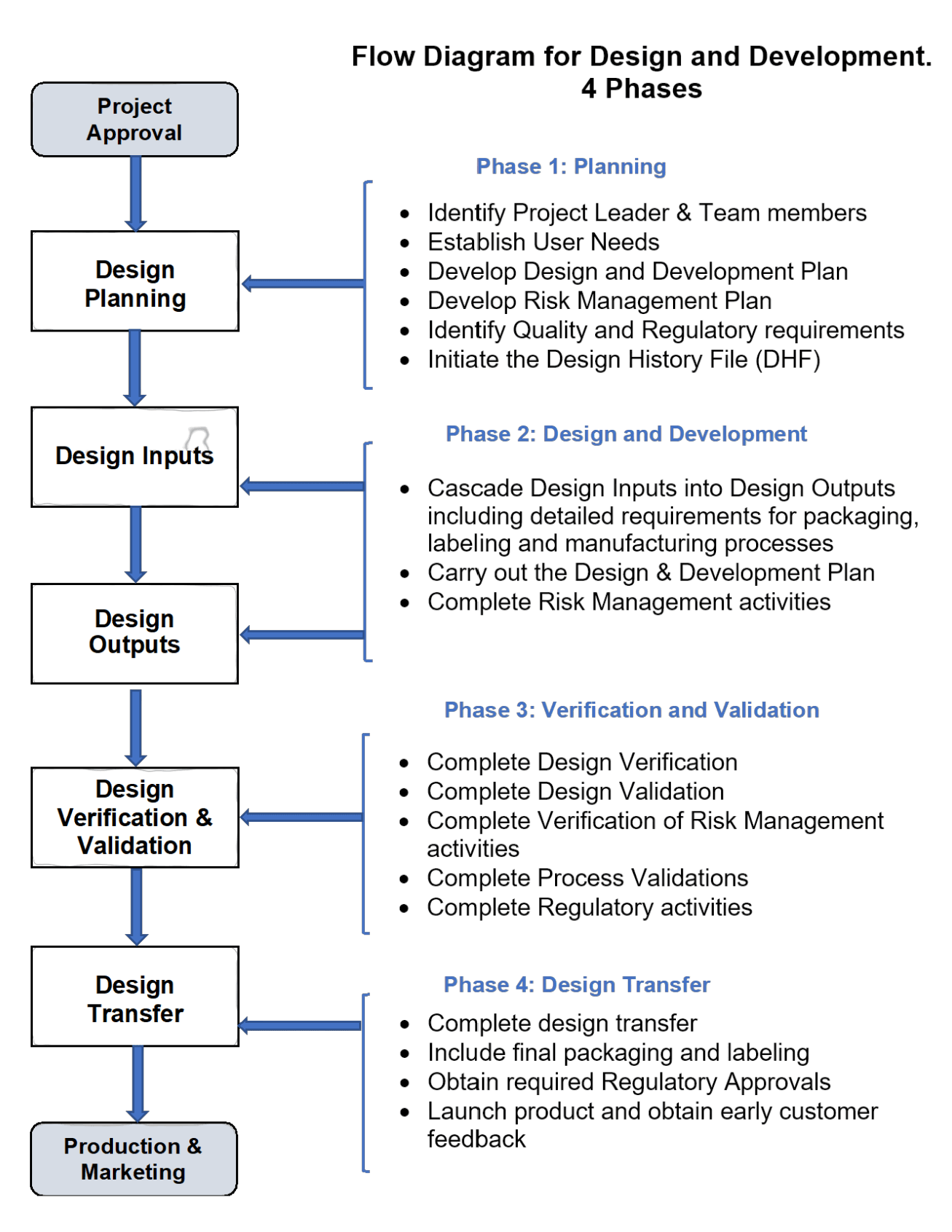
Before getting into more of my detailed guidance on each of the Design and Development process steps, see below for a process flow chart taken from our available procedure template SOP-731-01 Design and Development: If you would like to learn more on this SOP and other procedure we have available, just send me an email at: fasttrackiso13485@gmail.com
The 4 Phases of Design and Development
Not a specific regulatory requirement but I have found it good practice to define the Design and Development process into 4 main phases. Also to hold separate documented phase reviews and obtaining the appropriate approvals before moving onto to the next phase.
The diagram below shows these recommended phases along with the required design and development stages.
The 4 Phases are:
- Phase 1 Planning
- Phase 2 Design and Development
- Phase 3 Verification and Validation
- Phase 4 Design and Development Transfer
The intent of Design and Development Controls is to demonstrate that the medical device you have designed and built has been:
- Designed and developed to meet user needs and patient needs
- Designed to meet the design inputs and requirements
- Provide evidence that design outputs meet the inputs
- Meets safety and performance criteria
One other very important requirement and best practice is to incorporate throughout your design and development project Risk Management. Make sure you use the guidance for this by following the current ISO 14971 Medical devices ? Application of risk management to medical devices.
Risk Management is used to identify, evaluate, analyze, assess, and take appropriate risk reduction actions to mitigate those potential product issues.
Within Design Control the Risk Management activity has a strong correlation and having good Design Controls with related Risk Management practices will significantly improve the quality, safety, and the effectiveness of your medical device.
Now let?s review the Design and Development Stages:
Phase 1: Planning
So your medical device design and development project proposal has executive management approval and now it?s time to start the formal design project, and one following each of the ISO 13485:2016 quality system requirements.
A well-managed design and development project should have a formal documented plan which includes the following:
Selection of a Project Leader and team members:
The best practice that I have seen frequently, and have been part of, is the use of a cross functional team for Design and Development projects. It should not be a solo effort on the part of R&D. The full-time team members will most often be only from R&D with other department representatives getting involved as required. The size of the team and represented areas will depend on the complexity of the device design and may also require outside suppliers to be involved.
All team members need to be trained in the procedures and requirements for Design and Development.
The Project Leader who is normally from R&D Engineering is responsible for the creation and management of the design and development project plan including:
- Leading the design and development project
- Holding project design reviews and design phase reviews and obtaining approvals.
- Leading the project team consisting of, as appropriate, members from:
- Design Engineering
- Manufacturing Engineering
- Quality and Regulatory
- Manufacturing Operations
- Purchasing
- Marketing
- Other specialists as required
Establish User Needs:
The FDA says that design controls should start with User Needs, and that should be obvious, but sometimes unfortunately it is not always the case. It could be at a start-up medical device company the design project may begin with someone saying, ?hey let?s make a copy, or improvement to this current device and market it?, without asking for whom they are making it, and what the user needs.
So, understanding and documenting the User Needs is the right way to start a project and will meet the ISO 13485 and FDA Design and Development requirements. Also, when you get ready to determine the design validation plan it?s much easier to determine the validation protocols and acceptance criteria if the goals have been determined and documented.
When you?re establishing and documenting your User Needs here are some of the key questions to consider;
- What are the clinical needs or problems the device is required to solve?
- Who is going to be the device user?
- What are the key attributes and features that should be included in the devise design?
- How will the user use and interact with the device?
- Are there products that your device may interact with?
- What type of medical procedures will the device be used for?
- Is the device a single-use, disposable device, or is it a multi-use device?
- Where will the device be used?
One other important requirement to remember is that each User needs your document as part of your Design Inputs and the Design and Development Plan should be measurable. You will need during the verification and validation steps to verify that your final design addresses those User Needs,
Document the Design and Development Plan:
The overall documented Design and Development Plan will include all of the listed items here and for each stage of the project. The plan should be documented and be part of the Design History File and will be updated as each activity is carried out.
The Design and Development Plan should include the following:
- Identify Project Leader & Team members
- Establish User Needs
- Develop Risk Management Plan
- Identify Quality and Regulatory requirements
- Initiate the Design History File (DHF)
Develop Risk Management Plan:
Risk Management activities begin in the Planning phase and continue throughout the design process and the lifecycle of the product. In Phase 1, the Risk Management Plan is completed and documented.
As Phase 2 progresses the risk analysis is updated and should include: (Refer to ISO 14971 for Risk Management techniques.)
� Hazard Analysis and identification of possible hazards
� Calculation of risk, under normal and fault conditions
� Determination of risk acceptability
� Reduction of unacceptable risks to acceptable levels
� Include software risk analysis where appropriate
� Ensure that any changes do not introduce new hazards
Identify Quality and Regulatory requirements:
The Quality and Regulatory team member(s) will determine the Quality and Regulatory requirements with the details depending on the medical device classification, as well as the intended market, and regulatory requirements. This will be documented as part of the Project Plan.
Globally there are different agencies that govern and regulate medical devices as well as many requiring the ISO 13485:2016 Medical devices - Requirements for regulatory purposes. In the US there are the FDA requirements, Europe has the European Competent Authority, Canada has Health Canada, and so on.
The Quality and Regulatory project team members will need to understand, depending on where the medical device is planned to be sold, the regulations and requirements that are applicable, and document a summary of these as part of the Design and Development Plan.
It?s also important to remember these are not just requirements for the approval of the medical device, but also regulations for you to follow for Design Controls, during the product development process.
Initiate the Design History File:
The Design History File is where you keep all the documents for each phase of your Design and Development project. The right time to create this file and start using it is right now, during stage 1 and the building of the Design Plan. The Design History File needs to be well organized and kept up to date throughout the entire project and accessible to everyone on the project team.
A clear index of the design phases should make finding documents easy and provide the history and traceability that is expected and required. This is not just when the design project is in the process but after you launch your medical device and the DHF is audited by the FDA or other regulatory agencies.
Phase 2: Design and Development
Design and Development Inputs:
The first thing I would like to emphasize here is that terminology is important, and you and your team should get used to using the terms in the FDA regulation and the ISO 13485 Standard. This can sometimes be a transition especially when it comes to Design Inputs as many, including myself, are used to using alternate terms for defining the start of a design i.e.:
- Design requirements
- Product specifications
- Design inputs
- Customer design requirements
- Product definition
Being consistent with your terminology and your design and development documentation will ease any confusion and provide clear communication and understanding when it comes to your expected FDA inspections and ISO audits.
So, What are Design Inputs?
Design Inputs come from the User Requirements, and define performance criteria, functional requirements, safety and usability, as well as features of your medical device design.
Design Inputs also need to include the following:
- applicable regulatory requirements and standards depending on your intended markets
- any applicable outputs from risk management
- information and lessons learned from previous similar designs, as appropriate
- comparable competitive products as appropriate
Design Inputs must be clear and unambiguous and each able to be verified or validated. Also, they need to be reviewed and approved prior to the start of the formal design process.
It is best practice to consider how you will verify or validate each of these design inputs at the time you are establishing those inputs. It's referred to sometimes as ?simultaneous engineering? with which it can provide benefits including multidisciplinary collaboration and a fast-track design process.
Design and development outputs:
As your Design and Development project progresses you will start to develop your Design Outputs. These are the specifications for the device and provide the information for purchasing, manufacturing, and any service provision. It can be in the form of engineering drawings, specification sheets etc.
Each of the design Outputs needs to correspond and meet the documented and approved Design Inputs. The Design Outputs need to be traceable to each of the Design Inputs.
An example of a Design Output record form that can be used for a simple device is shown below.
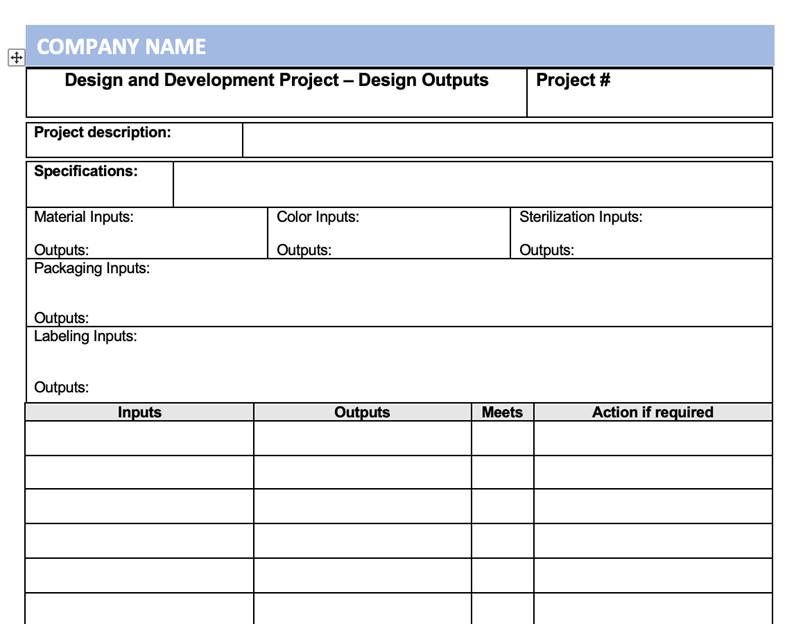
Design Outputs also need to be in a form suitable for verification against the Deign Inputs and need to be documented and approved before release.
Design and Development Review:
Medical Device companies need to consider design reviews, not as checking a box, but adding real value to help design a medical device that meets user needs and provides a successful design. Design Reviews are also a regulatory requirement to meet ISO 13485:2016 as well as FDA.
They are also a way to ensure and document the design and development process is on track and meets planned expectations. Design Reviews are conducted throughout the project as shown in the diagram below.
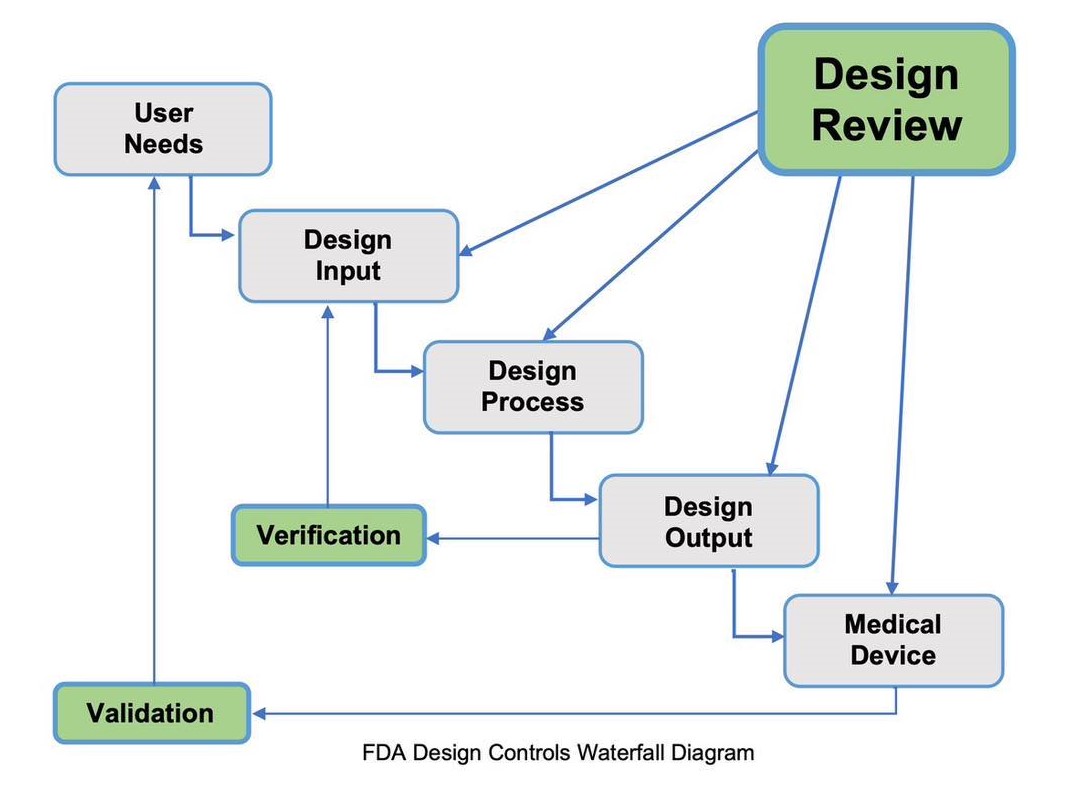
Phase 3: Verification and Validation
Design & development verification:
First, before we get into how you carry out design verification let's clarify any potential confusion between the meaning of verification versus validation. According to the FDA, they define them as follows:
Verification: Means confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.
Validation: Means confirmation by examination and provision of objective evidence that the particular requirements for specific intended use can be consistently fulfilled.
So as mentioned earlier, when you?re developing the Design Inputs you should have a good idea and plan for how to conduct the Design Verification. The basic goal is to prove through inspection and testing that the Design Outputs meet the Design Inputs.
Plan Design Verification and document before you begin the inspection and testing process.
Document the results and indicate pass or failure of requirements along with any required follow-up actions.
Design and development validation:
Design Validation is to confirm that the medical device you have designed consistently meets the design specifications, user needs, and intended uses. This means you should be involving the potential customers and end users to provide you with documented feedback on the design, and if it meets their needs.
Validation testing can be performed under actual or simulated conditions and must be performed using a product that was manufactured using final production equivalent documentation, materials, and processes. This also means that the manufacturing, testing, and inspection process need to have been completed, and as appropriate, all of the required Process Validations.
It's always best practice to have established early on in the project a Master Validation Plan, which would include all the required Process Validations and Design Validations. This MVP needs to include for the Design Validation activities, how many end users are required for the validation, what type of testing is to be performed, acceptance criteria, etc.
Design validation must be completed before the commercial distribution of the device.
????????????????????Phase 4: Design and Development Transfer
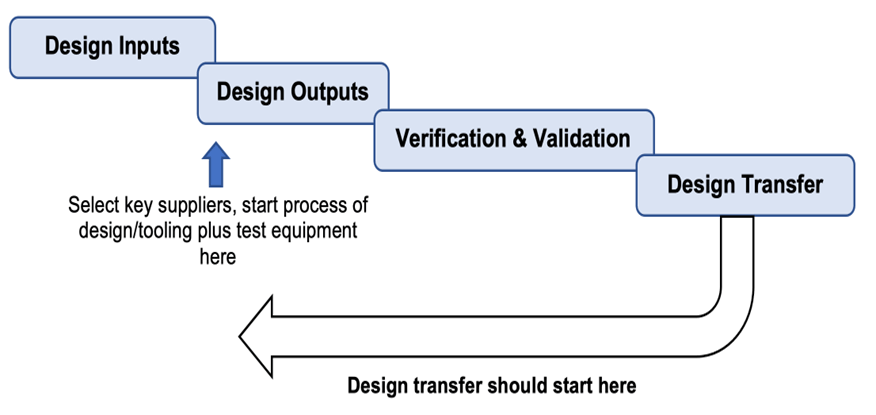
Design and development transfer is not a single event that occurs once a design is complete and validated but should start early in the design and development process.
As soon as any part of the design or manufacturing process is finalized, including verification and validation, it can be transferred to manufacturing.
As shown in the diagram below it is more of a simultaneous activity and can start as soon as parts of the design and manufacturing processes begin to become finalized.
For any company this approach makes sense and for a start-up medical device company this is critical for the fast-track approach.
Any time a partial design transfer takes place a design review should be held first, and the final transfer is also preceded by a design review and all documented.
Control of design and development changes
There will be changes you make to your medical device design as you develop your design, and these will be documented in your DHF.
Once the design is finalized and released through the Design Transfer process any new changes being made will be required to follow a documented design change procedure.
We review this in detail in a separate blog post and you can click on the link below where you can read or download the e-book:
Design and Development File:
The Design and Development File, or Design History File (DHF), which it is commonly referred to, has been updated continually as the team works its way through the product design and development.
It contains or references records generated that demonstrate the products conformity to design and development requirements and any changes that were made. The file(s) will include documents and records for the following:
- User needs and Design Inputs
- Design Outputs
- Design Reviews
- Design Verification and Validation
- Design Transfer
- Design Changes
Keep in mind throughout your project that your DHF is very likely to be audited by FDA and ISO auditors, so take your time to ensure your files are complete with all the required documentation and they are well organized. It can make a very significant difference on how the audits go, trust me on this one.

Document Control of Design Files
Keeping control of Design Files can be a major challenge and making sure they are complete with all the records and ready to be audited by FDA or ISO auditors can be a real headache.
My own experience many times, I can recall for a regulatory audit, and just for one design and development project, the requested files being wheeled into the audit room on a 4 wheeled trolley. We had great confidence in our doc control practices but it still made me anxious.
In today?s world, there is a better alternative, and we can recommend one of our partners of Fast-Track QMS Consultants, who offer an easy-to-use, cloud-based software platform specifically designed to help medical device companies get to market faster by better managing their Design Controls.
To learn more, and for a free demo click here: link
Final Thoughts:
Design Transfer completed and the Design History File updated and complete. This will include completion of your Regulatory Plan and you should have by all the required regulatory clearances, depending on your markets.
Regulatory requirements such as FDA 510(k) approvals, European CE Marking and Technical Files can be time consuming taking many months. The submission process can start after completing the verification process and you?re into the validation plan, but this also depends on the classification of your device.
So now you?re ready to launch your product.
One best practice I have seen is to have a controlled launch plan that limits the early release of the new medical device to a few selected distributors. This is in order to obtain Early Warning of any issues.
This can be especially beneficial for a start-up medical device company that may want to test out their distribution, feedback and customer complaint procedures, as well of course confirm the customer satisfaction and performance of the device.
This Early Warning Program can involve incentives to those selected distributors and/or customers as well as a regular follow-up routine, rather than just waiting for issues to be reported.
So, hope you have found this guide to be useful and the systematic step-by-step process described for Design and Development of your Medical Device is not just about meeting the ISO 13485:2016 Standard and FDA requirements but is absolutely the best practice for designing and developing a quality and safe medical device.
How can Fast-Track QMS Consultants help?
Want to learn more about Fast-Track QMS Consulting and the document bundles and consulting services we can provide, including a proven, Design and Development, procedure and supporting templates.
??
We can also offer related procedures for Design and Development including; Control of Design and Development Changes, Risk Management, and Device Master Record.